Navigation
Documentation
This page contains explanations and references for the tools provided.
Licensing & How To Cite Oligowizard
All tools available here are free to be used for personal, academic and commercial use under Creative Commons licensing.
For more information please refer to the End User Licence Agreement or get in touch.
We kindly ask you to cite our resource - you may adapt the following sentence:
'Oligo properties were calculated using the oligowizard nucleic acid toolbox - available at https://oligowizard.com'
Recent Patchnotes
2026-01-22 - Version 4.2.1- Preparations ahead of version 5.0.0
- Minor style and quality of life improvements
2025-04-20 - Version 4.2.0
- Released Oligowizard premium!
- Released API interface for oligo calculations (github)
- Added Confidential mode for premium users
- Vector Graphics Tool now comes with Templates
- Added yield calculation to /advanced
- Responsive Design for mobile and tablets
- Re-worked full documentation (see below)
- Minor style and quality of life improvements
2025-03-30 - Version 4.1.0
- Added more customisation to vector graphic drawing
- Fixed bug where tM calculations would not account for concentration
- Added Captcha for signup and login
- Minor style and quality of life improvements
2024-11-18 - Version 4.0.3
- Dedicated Structure drawing feature
- misc. design changes
- misc. quality of life improvements
2024-10-24 - Version 4.0.2
- Fixed bug for tm calculation at low salt
- Fixed bug for oligos with no extinction at 260 nm
- misc. bug fixes
2024-09-30 - Version 4.0.1
- misc. hotfixes
2024-09-29 - Version 4.0.0
- Major release of a new Version!
- Re-design of the user interface
- Added chemical structure (*.cdxml) drawing tool
- Improved HPLC trace analysis tool and added analytical features
- Completely overhauled batch processing
- Increased number of favourites from 5 to 10
- Custom Nucleotides now account for molar extinction
- significant improvements to the backend architecture
- misc. security improvements
Oligo Properties Calculator
The Oligowizard advanced calculator provides an intuitive interface to calculate biophysical properties of nucleic acid sequences. The tool is able to process modified nucleic acids and mixmers with common 2’ Ribose modifications as well as both PO/PS backbones.
INPUT
- Sequence (sequence) [STRING] required
- A260 (A260) [FLOAT] optional, default = 1
- Salt Concentration mM Na+ (Na_conc) [FLOAT] optional, default = 50
- Salt Concentration mM K+ (K_conc) [FLOAT] optional, default = 0
- Salt Concentration mM Mg2+ (Mg_conc) [FLOAT] optional, default = 0
- 3'-Modification (three_prime) [STRING]/[FLOAT] optional, default = "OH"
- 5'-Modification (five_prime) [STRING]/[FLOAT] optional, default = "OH"
- 5' Modification was attached via PS linkage (five_is_PS) [BOOL] optional, default = "FALSE"
Sequences should be in 5' to 3' direction. Oligowizard uses a dedicated code to represent the most commonly used 2' modification patterns: DNA: (A / C / G / T), RNA: ( D / E / F / U), LNA: (H / I* / J / K), MOE: (L / M* / N / O), OMe: (P / Q / R / S), 2'- F:(V / W / X / Y) Note here, that bases indicated with an asterisk (*) carry an 5-Me on the nucleobase, as these are the most commonly used version of the phosphoramidite. Lower case letters are used to denote phosphorothioate linkages (PS) on the backbone, while capital letters symbolise regular phosphate diesters (PO). Placement of the PS linkage is relative to the 3' postion of the nucleotide. AC*GT == AcGT. If the character at the 3' end of the sequence is in lower case, no changes are made to the result, as the 3' position does not usually carry a phosphate group. This distinction is however important, if a modification is attached to the 3' end!
The measured absorbance (optical densitiy) at 260 nm. The value is used to calculate concentration, and melting temperature.
The concentration of sodium cations. Value is used in the melting temperature calculation
The concentration of potassium cations. Value is used in the melting temperature calculation
The concentration of magnesium cations. Value is used in the melting temperature calculation
Modification attached at the three prime position of the oligonucleotide. If a string is passed, a lookup table for predefined modifications will be used to load mass, absorbance / melting properties, possible protecting groups / oxidation states. If a floating point number is passed, mass calculations will use this (ommitting the 3'Oxygen) -- no further assumptions will be made about absorbance or melting properties. Linkage type (PO/PS) between the three prime nucleotide and possible modifcation will be calculated based on the capitalisation of the last character in the sequence.
Modification attached at the five prime position of the oligonucleotide. If a string is passed, a lookup table for predefined modifications will be used to load mass, absorbance / melting properties, possible protecting groups / oxidation states. If a floating point number is passed, mass calculations will use this (ommitting the 3'Oxygen) -- no further assumptions will be made about absorbance or melting properties.
Used to determine the mass.
OUTPUT
- Sequence (sequence) [STRING]
- Custom NT removed (sequence_sanitised) [STRING]
- DNA-PO eq. (DNAeq) [STRING]
- 5' Modification (five_prime) [STRING]
- 3' Modification (three_prime) [STRING]
- Absorbance at 260 nm (A260) [FLOAT]
- Length (oligo_length) [INTEGER]
- GC Content (gc_cont) [FLOAT]
- Reverse Complement (rev_comp) [STRING]
- DNA melting temperature (tm1) [FLOAT]
- Melting temperature approximation (tm2) [STRING]
- Molecular weight (canonical) (mass1) [FLOAT]
- Molecular weight - Alternative 3' Mass (mass2) [FLOAT]
- Alternative 3' Mass Identity (mass2_text) [STRING]
- Molecular weight - Alternative 5' Mass (mass3) [FLOAT]
- Alternative 5' Mass Identity (mass3_text) [STRING]
- Molecular weight - Double modified (mass4) [FLOAT]
- Alternative Mass Identity (mass4_text) [STRING]
- Molar Extinction Coefficient (simple) (molext) [FLOAT]
- Concentration (Simple extinction) (conc1) [FLOAT]
- Concentration (Simple extinction) (conc2) [FLOAT]
- Molar Extinction Coefficient (NN Model) (molext_nn) [FLOAT]
- Concentration (NN Model) (conc1_nn) [FLOAT]
- Concentration (NN Model) (conc2_nn) [FLOAT]
Returns the input sequence.
Returns the input sequence with any custom nucleotides replaced by the closest mapping DNA base (based on user setting).
Returns the DNA-Phosphodiester equivalent of the input sequence: as capital letters (PO) with any non-DNA bases replaced by the corresponding DNA bases.
Returns the input 5' modification mapped to its full name, or entered FLOAT value as string.
Returns the input 3' modification mapped to its full name, or entered FLOAT value as string.
Returns the input absorbance at 260 nanometer.
Returns the length of the nucleotide in nucleotides (not counting terminal modifcations).
Returns the percentage of Guanosine and Cytosin bases (or equivalents thereof) in the sequence.
Reverse complement of the sequence. Custom nucleotides will be replaced according to user-specified equivalent bases. Notably: as all characters will be inverted in capitalisation and backbone identity is relative to the 3' linkage, postion of PS linkages are not preserved correctly (AcGGT-> ACCgT == A-C*G-G-T -> A-C-C-G*T) this is a known bug.
Melting temperature, assuming the oligonucleotide is a DNA PO strand [1][2][3]
Estimated melting temperature with correction values applied for sugar modifications where available (currently, MOE and LNA). Returned as a string with a range. Takes user-specified modifiers into account.
Molecular weight of the oligo and terminal modifications with all protecting groups removed (DMT-OFF)
If the modification at the 3' position of the oligo has an alternative mass depending on protection groups / oxidative state, the corresponding mass is returned here.
Short explanation, which alternative form of the 3' modification was taken into account for the alternative mass.
If the modification at the 5' position of the oligo has an alternative mass depending on protection groups / oxidative state, the corresponding mass is returned here. If no modification was specified, the DMT-ON weight will be given here.
Short explanation, which alternative form of the 5' modification was taken into account for the alternative mass. For unmodified oligos, this will be "5' DMT protected"
If the modification at the 3' and 5' positions of the oligo have an alternative mass depending on protection groups / oxidative state, the corresponding mass is returned here. If no modification was specified for the 5' end, the DMT-ON weight will be given here in combination with the 3' alternative mass.
Short explanation, which alternative forms of the 3' and 5' modification was taken into account for the alternative mass.
Molar extinction coefficient (in L mol-1 cm-1) of the oligo and any present modifcations based on a simple extinction model. Will take user specified extinction values into account
Concentration of the oligo (in micromole per liter) based on simple extinction coefficient and specified absorbance at 260 nm (A260).
Concentration of the oligo (in nanogram per microliter) based on simple extinction coefficient and specified absorbance at 260 nm (A260).
Molar extinction coefficient (in L mol-1 cm-1) of the oligo based on the nearast neighbor model. Currently only takes nucleobases into account.
Concentration of the oligo (in micromole per liter) based on the nearast neighbor model extinction coefficient and specified absorbance at 260 nm (A260).
Concentration of the oligo (in nanogram per microliter) based on the nearast neighbor model extinction coefficient and specified absorbance at 260 nm (A260).
REFERENCES
[1] SantLucia et al. (1998): 'A unified view of polymer, dumbbell, and oligonucleotide DNA nearest-neighbor thermodynamics'[2] SantLucia et al. (1996): 'Improved Nearest-Neighbor Parameters for Predicting DNA Duplex Stability'
[3] Owczarzy et al. (2004): 'Effects of Sodium Ions on DNA Duplex Oligomers: Improved Predictions of Melting Temperatures'
Custom Nucleotides
- Cutom Building Blocks (custom_nt) [ARRAY[ARRAY]] optional, default = NULL
- Name (name) [STRING] required, default = "Custom Nucleotide"
- Molecular Weight (mass) [FLOAT] required, default = 0.0000
- Molar Extinction (extinction) [FLOAT] required, default = 0.0000
- Most closely resembles (ACGT) [CHAR[A,C,G,T]] required, default = "A"
- Effect on Melting Temperature (tm) [FLOAT](signed) required, default = 0.0000
- Colour (colour) [STRING] required, default = "#566573"
If you have an oligowizard.com account, you can define your own nucleotides using the numbers 0-9. You can specify these under settings. All [FLOAT] round to 4 decimal points
Name to identify the custom nucleotide
Chemical Structure Generation
This tool allows you to generate structure files (as *.cdxml) from a given nucleotide sequence. These files can be viewed and edited in a compatible third-party structure editor such as ChemDraw or ACD/ChemSketch.
INPUT
- Sequence (sequence) [STRING] required
- Bond Line Width (width) [INTEGER] optional, default = 1
- Atom Label Font Size (size) [INTEGER] optional, default = 12
- LabelFace (face) [INTEGER] optional, default = 96
- Scaling factor (scale) [FLOAT] optional, default = 0.45
Sequences should be in 5' to 3' direction. Oligowizard uses a dedicated code to represent the most commonly used 2' modification patterns: DNA: (A / C / G / T), RNA: ( D / E / F / U), LNA: (H / I* / J / K), MOE: (L / M* / N / O), OMe: (P / Q / R / S), 2'- F:(V / W / X / Y) Note here, that bases indicated with an asteriks (*) carry an 5-Me on the nucleobase, as these are the most commonly used version of the phosphoramidite. Lower case letters are used to denote phosphorothioate linkages (PS) on the backbone, while capital letters symbolise regular phosphate diesters (PO). Placement of the PS linkage is relative to the 3' postion of the nucleotide. AC*GT == AcGT. If the character at the 3' end of the sequence is in lower case, no changes are made to the result, as the 3' position does not usually carry a phosphate group.
Sets the width of the bonds.
Sets the font size for atom labels.
Determines the appearnces of atom labels - default/bold. Enter 97 for bold
Scales the size of the molecule. Especially useful to fit larger molecules on one page.
OUTPUT
- structure_XXXXXX.cdxml (outfile) [FILE]
File containing the oligonucleotide structure in cdxml format.
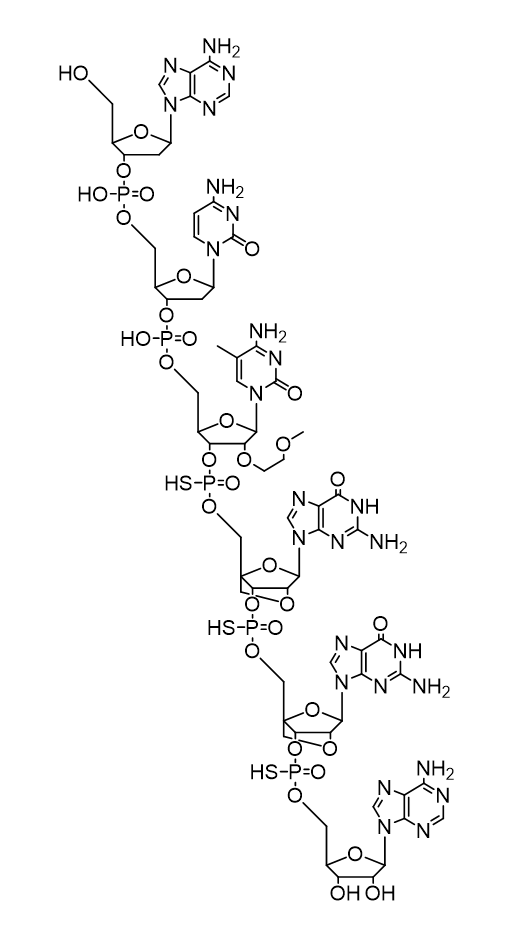
Example Chemical Structure
Disclaimer: ‘ChemDraw’ is a registered property of Revvity, Inc. (formerly PerkinElmer, Inc.), and ‘ACD/ChemSketch’ is a registered property of Advanced Chemistry Development, Inc. (ACD/Labs). OLIGOWIZARD LTD has no affiliation with Revvity, Inc. or ACD/Labs. The chemical structure drawing feature on oligowizard.com generates CDXML files compatible with software like ChemDraw (a separate licence may be required) and similar tools, including ACD/ChemSketch. OLIGOWIZARD LTD does not provide software licences or support for these third-party applications.
Vector Graphics Drawing Tool
Tool to generate simple vector graphics of nucleic acid chains
INPUT
- Sequence (sequence) [STRING] required
- Settings (/settings) [ARRAY] required
- Border / Stroke Colour (stroke_colour) [STRING] required, default = "#212f3d"
- Stroke Width (stroke_width) [INTEGER] required, default = "0"
- Circle Radius (circle_radius) [INTEGER] required, default = 60
- Backbone Thickness (backbone_thickness) [INTEGER] required, default = 50
- Font Colour (font_colour) [STRING] required, default = "black"
- Font Size (font_size) [STRING] required, default = "110"
- Font Weight (font_weight) [STRING] required, default = "bold"
- DNA Colour (colour_DNA) [STRING] required, default = "#566573"
- RNA Colour (colour_RNA) [STRING] required, default = "#3498db"
- MOE Colour (colour_MOE) [STRING] required, default = "#9b59b6"
- Methoxy (OMe) Colour (colour_OMe) [STRING] required, default = "#138d75"
- LNA Colour (colour_LNA) [STRING] required, default = "#ec7063"
- 2'-F Colour (colour_F) [STRING] required, default = "#eb984e"
- Phosphate Backbone (PO) (colour_PO) [STRING] required, default = "#212f3d"
- Phosphothioate Backbone (PS) (colour_PS) [STRING] required, default = "#c0392b"
- Load a Preset (preset) [STRING] optional, default = NULL
Sequences should be in 5' to 3' direction. Oligowizard uses a dedicated code to represent the most commonly used 2' modification patterns: DNA: (A / C / G / T), RNA: ( D / E / F / U), LNA: (H / I / J / K), MOE: (L / M / N / O), OMe: (P / Q / R / S), 2'- F:(V / W / X / Y) Lower case letters are used to denote phosphorothioate linkages (PS) on the backbone, while capital letters symbolise regular phosphate diesters (PO). Placement of the PS linkage is relative to the 3' postion of the nucleotide. AC*GT == AcGT. If the character at the 3' end of the sequence is in lower case, no changes are made to the result, as the 3' position does not carry a phosphate group.
The settings menu allows you to customise the style of the vector graphic (login required).
Defines the colour of outline strokes.
Sets the thickness of outline strokes.
Radius of each nucleotide circle.
Thickness/height of the backbone line.
Sets the colour of base labels.
Size of base label text.
Controls label thickness (normal, bold).
Defines the colour of DNA monomers.
Defines the colour of RNA monomers.
Defines the colour of MOE monomers.
Defines the colour of OMe monomers.
Defines the colour of LNA monomers.
Defines the colour of 2'-F monomers.
Defines the colour of PO backbone.
Defines the colour of PS backbone.
Loads and applies a preset for oligo vector draw
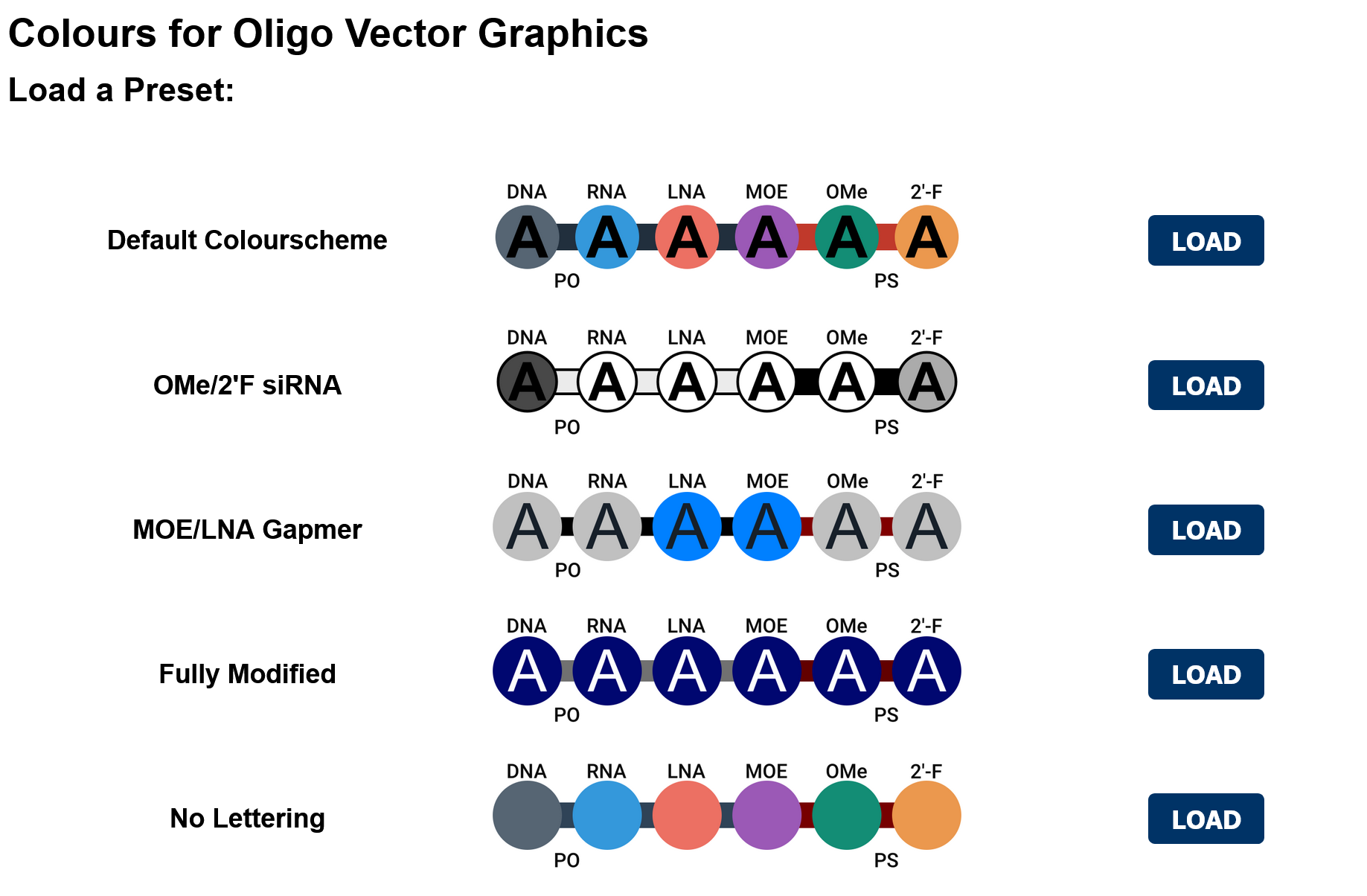
Available Colour Presets
In addition to the customisable settings, we also provide a suite of ready-to-go presets for common types of nucleic acid therapeutics. Presets can be loaded by the click of a button and then further customised with the settings.
OUTPUT
- oligovector_XXXXXX.svg (outfile1) [FILE]
- oligolegend_XXXXXX.svg (outfile2) [FILE]
The tool returns a vector graphic of the drawn oligo chain. The figure can be viewed and edited in third-party vector graphic software (such as adobe illustrator or Inkscape)

Dynamically generated legend for the oligo draw feature
The tool automatically generates a second vector graphic containing a figure legend. The legend can be viewed and edited in third-party vector graphic software (such as adobe illustrator or Inkscape)
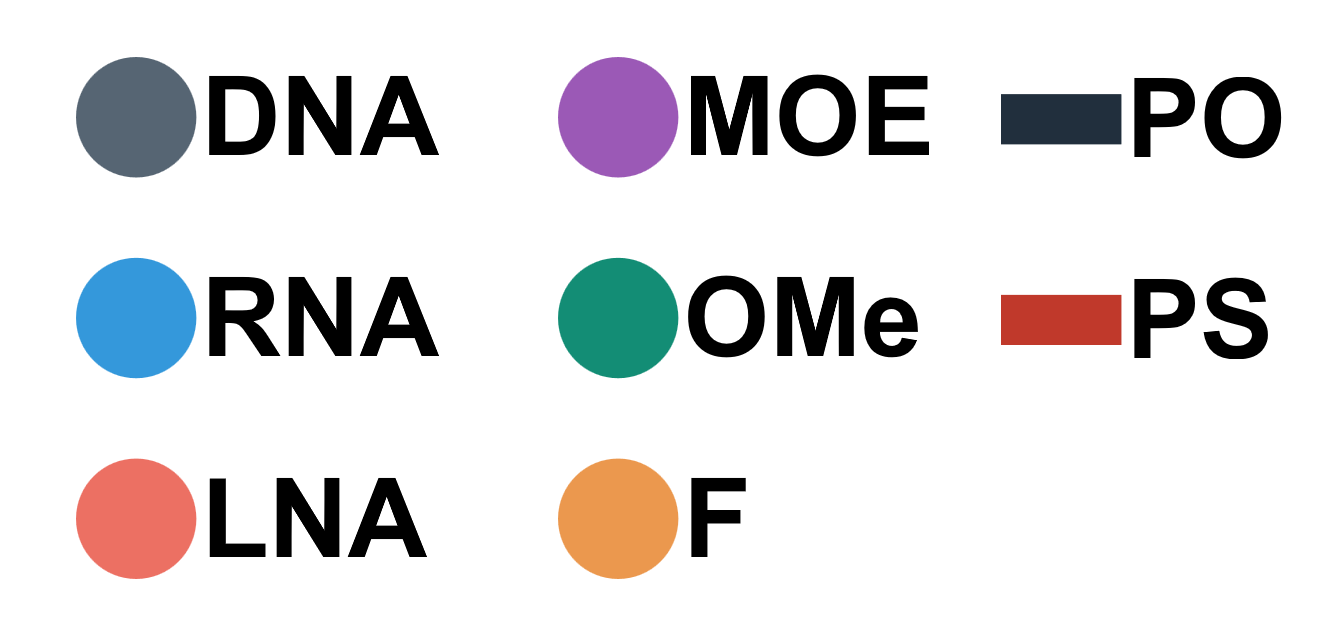
Dynamically generated legend for the oligo draw feature
Batch Calculator
Tool to perform Oligo Property calculations on a library of compounds at the same time. Tje tool works similar to the calculator interface, but is optimised to handle large files rather than manual inputs. The tool returns a table that can be directly viewed in the browser or downloaded. Customisation of the calculation is limited, if more tailored high-throughput calcuations are required, we recommend using the calculator via our API instead.
INPUT
- Upload new file (filename) [FILE] required
- Name (name) [STRING] required
- Sequence (sequence) [STRING] required
- 5'-Modification (five_prime) [STRING]/[FLOAT] required, default = "OH"
- 5' Modification was attached via PS linkage (five_is_PS) [BOOL] required, default = "FALSE"
- 3'-Modification (three_prime) [STRING]/[FLOAT] required, default = "OH"
- A260 (A260) [FLOAT] required, default = 1
- Select Parameters to Calculate (filename) [ARRAY[BOOL]] required
Data is entered by uploading a comma seperated
To ensure compatibility, please exclusivle use the template provided here :
[DOWNLOAD TEMPLATE]
When saving, do not change the file type (*.csv) and make sure the file does not contain any special characters!
Please avoid special characters, use the decimal point '.' to denote decimal values (rather than a comma ',') and make sure all columns are filled.
Batch mode will perform melting temperature calculations assuming 50 mM sodium cations.
Please note, that in contrast to the UI based calculator, all input fields are required! Empty cells will cause the script to fail.
Name / ID of the compound. Used to identify the row in the output.
Sequences should be in 5' to 3' direction. Oligowizard uses a dedicated code to represent the most commonly used 2' modification patterns: DNA: (A / C / G / T), RNA: ( D / E / F / U), LNA: (H / I* / J / K), MOE: (L / M* / N / O), OMe: (P / Q / R / S), 2'- F:(V / W / X / Y) Note here, that bases indicated with an asteriks (*) carry an 5-Me on the nucleobase, as these are the most commonly used version of the phosphoramidite. Lower case letters are used to denote phosphorothioate linkages (PS) on the backbone, while capital letters symbolise regular phosphate diesters (PO). Placement of the PS linkage is relative to the 3' postion of the nucleotide. AC*GT == AcGT. If the character at the 3' end of the sequence is in lower case, no changes are made to the result, as the 3' position does not usually carry a phosphate group. This distinction is however important, if a modification is attached to the 3' end!
Modification attached at the five prime position of the oligonucleotide. If a string is passed, a lookup table for predefined modifications will be used to load mass, absorbance / melting properties, possible protecting groups / oxidation states. If a floating point number is passed, mass calculations will use this (ommitting the 3'Oxygen) -- no further assumptions will be made about absorbance or melting properties.
Used to determine the mass.
Modification attached at the three prime position of the oligonucleotide. If a string is passed, a lookup table for predefined modifications will be used to load mass, absorbance / melting properties, possible protecting groups / oxidation states. If a floating point number is passed, mass calculations will use this (ommitting the 3'Oxygen) -- no further assumptions will be made about absorbance or melting properties. Linkage type (PO/PS) between the three prime nucleotide and possible modifcation will be calculated based on the capitalisation of the last character in the sequence.
The measured absorbance (optical densitiy) at 260 nm. The value is used to calculate concentration, and melting temperature.
Array of values to be calculated - see below.
OUTPUT
- batch_XXXXXX.csv (outfile) [FILE]
- Name (name) [STRING]
- Sequence (sequence) [STRING]
- 5' Modification (five_prime) [STRING]
- 3' Modification (three_prime) [STRING]
- Absorbance at 260 nm (A260) [FLOAT]
- Length (oligo_length) [INTEGER]
- GC Content (gc_cont) [FLOAT]
- Reverse Complement (rev_comp) [STRING]
- DNA melting temperature (tm) [FLOAT]
- Molecular weight (canonical) (mass1) [FLOAT]
- Molar Extinction Coefficient (simple) (molext) [FLOAT]
- Concentration (Simple extinction) (conc1) [FLOAT]
- Concentration (Simple extinction) (conc2) [FLOAT]
- Molar Extinction Coefficient (NN Model) (molext_nn) [FLOAT]
- Concentration (NN Model) (conc1_nn) [FLOAT]
- Concentration (NN Model) (conc2_nn) [FLOAT]
- Molecular weight - Alternative 3' Mass (mass2) [FLOAT]
- Alternative 3' Mass Identity (mass2_text) [STRING]
- Molecular weight - Alternative 5' Mass (mass3) [FLOAT]
- Alternative 5' Mass Identity (mass3_text) [STRING]
- Molecular weight - Double modified (mass4) [FLOAT]
- Alternative Mass Identity (mass4_text) [STRING]
Comma separated table containing the results of the calculation.
Name / ID of the compound. Used to identify the row in the output.
Returns the input sequence.
Returns the input 5' modification mapped to its full name, or entered FLOAT value as string.
Returns the input 3' modification mapped to its full name, or entered FLOAT value as string.
Returns the input absorbance at 260 nanometer.
Returns the length of the nucleotide in nucleotides (not counting terminal modifcations).
Returns the percentage of Guanosine and Cytosin bases (or equivalents thereof) in the sequence.
Reverse complement of the sequence. Custom nucleotides will be replaced according to user-specified equivalent bases. Notably: as all characters will be inverted in capitalisation and backbone identity is relative to the 3' linkage, postion of PS linkages are not preserved correctly (AcGGT-> ACCgT == A-C*G-G-T -> A-C-C-G*T) this is a known bug.
Melting temperature, assuming the oligonucleotide is a DNA PO strand [1][2][3]
Molecular weight of the oligo and terminal modifications with all protecting groups removed (DMT-OFF)
Molar extinction coefficient (in L mol-1 cm-1) of the oligo and any present modifcations based on a simple extinction model. Will take user specified extinction values into account
Concentration of the oligo (in micromole per liter) based on simple extinction coefficient and specified absorbance at 260 nm (A260).
Concentration of the oligo (in nanogram per microliter) based on simple extinction coefficient and specified absorbance at 260 nm (A260).
Molar extinction coefficient (in L mol-1 cm-1) of the oligo based on the nearast neighbor model. Currently only takes nucleobases into account.
Concentration of the oligo (in micromole per liter) based on the nearast neighbor model extinction coefficient and specified absorbance at 260 nm (A260).
Concentration of the oligo (in nanogram per microliter) based on the nearast neighbor model extinction coefficient and specified absorbance at 260 nm (A260).
If the modification at the 3' position of the oligo has an alternative mass depending on protection groups / oxidative state, the corresponding mass is returned here.
Short explanation, which alternative form of the 3' modification was taken into account for the alternative mass.
If the modification at the 5' position of the oligo has an alternative mass depending on protection groups / oxidative state, the corresponding mass is returned here. If no modification was specified, the DMT-ON weight will be given here.
Short explanation, which alternative form of the 5' modification was taken into account for the alternative mass. For unmodified oligos, this will be "5' DMT protected"
If the modification at the 3' and 5' positions of the oligo have an alternative mass depending on protection groups / oxidative state, the corresponding mass is returned here. If no modification was specified for the 5' end, the DMT-ON weight will be given here in combination with the 3' alternative mass.
Short explanation, which alternative forms of the 3' and 5' modification was taken into account for the alternative mass.
HPLC Toolbox
The oligowizard HPLC tool allows you to visualise and quantify simple HPLC traces using exported data from liquid chromatography systems.
INPUT
- Upload new file (filename) [FILE] required
- Canvas Title (title) [STRING] optional, default = "New HPLC Trace"
- Title font size (title_fontsize) [INTEGER] optional, default = 42
- Canvas Width (canvasx) [INTEGER] optional, default = 1000
- Canvas Height (canvasy) [INTEGER] optional, default = 300
- Axis Font Size (axis_fontsize) [INTEGER] optional, default = 24
- X-Axis Label (xaxis_label) [STRING] optional, default = "Retention time [min]"
- X-Axis Major Tick Mark (xaxis_major) [FLOAT] optional, default = 1
- X-Axis Minor Tick Mark (xaxis_minor) [FLOAT] optional, default = 0.2
- X-Axis Start (min_minutes) [FLOAT] optional, default = 0
- X-Axis Stop (max_minutes) [FLOAT] optional, default = 15
- Y-Axis Label (yaxis_label) [STRING] optional, default = "A260 [mAU]"
- Y-Axis Limit (limit) [INTEGER] optional, default = 3000
- Y-Axis Major Tick Mark (yaxis_major) [FLOAT] optional, default = 1000
- Y-Axis Minor Tick Mark (yaxis_minor) [FLOAT] optional, default = 100
- Trace Thickness (stroke) [FLOAT] optional, default = 3
- Trace Colour (colour) [STRING] optional, default = "#0000FF"
- Analysis (analysis) [STRING] optional, default = "none"
- Analysis Sensitivity (sensitivity) [INTEGER] optional, default = 30
- Analysis Peak Font Size (peak_fontsize) [INTEGER] optional, default = 18
The tool was developed using *.csv exports from the agilent chemstation but will accept any dataset of x = time, y = signal that is comma or tab seperated.
Title to be displayed at the top of the image. This will often be the name/ID of the analysed sample.
Font size for the canvas title.
Width in pixels of the generated image.
Height in pixels of the generated image.
Font size for the x and y-axis labels.
Label for the x-axis of the graph - retention time.
Interval in minutes for the larger tick marks on the x-axis
Interval in minutes for the smaller tick marks on the x-axis
Starting time in minutes for the x-axis. Can be used to zoom in / crop a particular interval of the trace. We recomend to leave the value at 0.
Stopping time in minutes for the x-axis. Can be used to zoom in / crop a particular interval of the trace. If left empty, the script will auto scale the axis to the full dataset (recommended).
Label for the y-axis of the graph - absorbance signal.
Max value for the y-axis (signal). If left empty, the script will autoscale based on the dataset (recommended).
Interval for the larger tick marks on the y-axis
Interval for the smaller tick marks on the y-axis
Stroke width for the path of the HPLC trace.
Stroke colour for the path of the HPLC trace.
Selector for the type of analysis ["none"/"peaks"/"purity"]. ["none"]: No analysis will be performed - the default. ["peaks"]: A peak detection algorithm will be run on the dataset. Peak detection is performed in four steps: delta from the baseline, first derivative of the signal (to identify the apex of the peak), and second derivative (to distuingush shoulders and overlapping signals), symmetry of the detected peak. Notably, the peak detection algorithm is still under development and might not accuratly detect poorly resolved or low signal-to-noise shoulders, or closely overlapping peaks. In the graphic, detected peaks will be indicated by their retention time. ["purity"]: A peak detection algorithm will be run on the dataset. Detected signals will then be integrated using their area under the curve. Peaks will be indicated in the trace with their retention time - different species will be indicated on the x-axis by red vertical bars. A table given retention time and calculated UV purity (based on AUC) will be displayed at the bottom of the graph. Please note: the peak table will only include species the algorithm considers to be true peaks (based on symmetry of the signal). Noisy background or highly asmmyetric peaks will be considered for purity calcuations only but not listed as a detected species in the table.
Determines the sensitivity of the peak detection algorithm (see above). The number given determines the delta between signal and background before peak detection is activated. A lower value will make the tool more sensitive. Please note: the baseline detection algorithm used is still under development, traces with low signal to noise ratio or high fluctutations in the baseline at the beginning of the trace will negatively affect the quality of the analysis.
Font size of the peak calling on the graph. If only analysis is desired, 0 can be chosen for the font size.
OUTPUT
- trace_XXXXXX.svg (outfile) [FILE]
Vector graphic file containing the HPLC trace (and analysis table). Files can be viewed and edited in third-party vector graphic software (such as adobe illustrator or Inkscape)
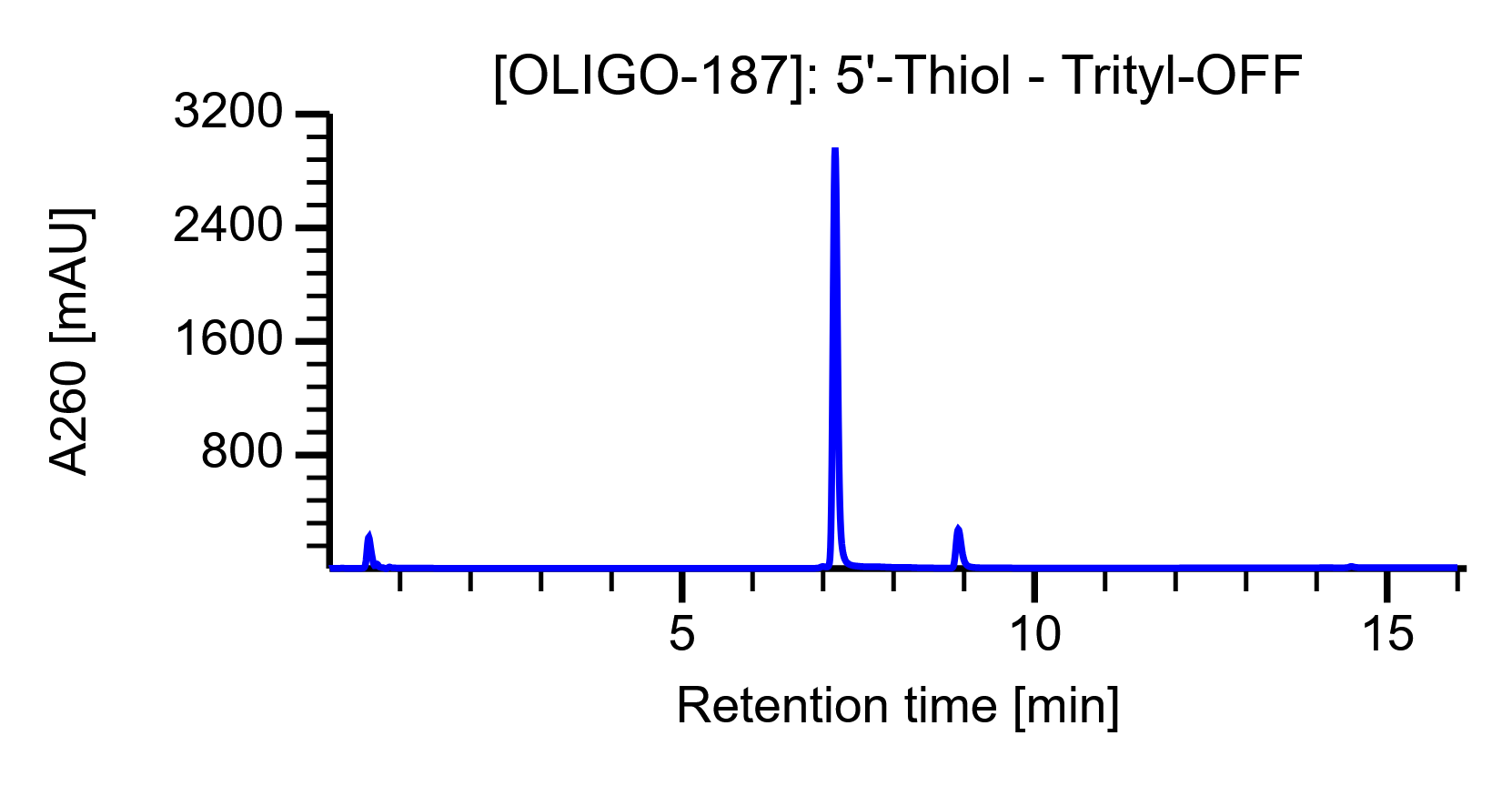
Example HPLC trace
The tool returns the HPLC trace as a graph: signal [mAU] is plotted over time [minutes] in blue.
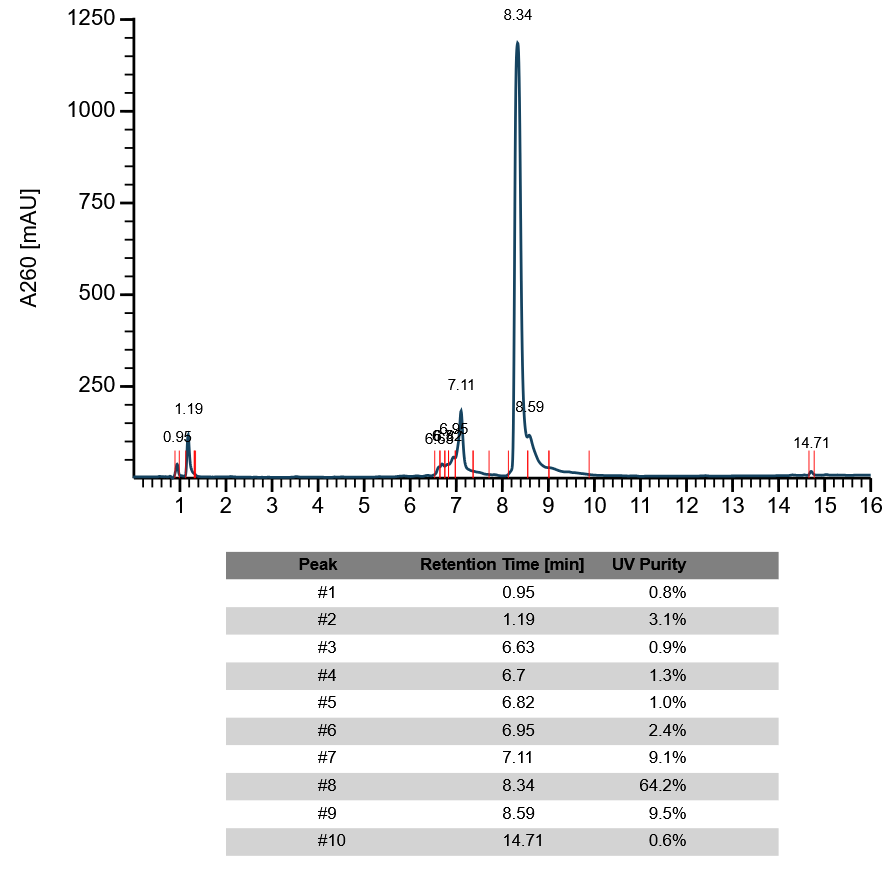
Example HPLC trace with analysis
The tool returns the HPLC trace as a graph: signal [mAU] is plotted over time [minutes] in dark grey.
Plasmid Map Drawing Feature
A simple tool to draw minimalistic Plasmid Maps and return vector graphics.
INPUT
- Upload new file (filename) [FILE] required
Data is entered by uploading a comma seperated text file.
Please use the provided template to ensure compatibility:
1st line: Plasmid Name[STRING]
2nd line: Length in Base Pairs (BP)[INTEGER]
Any other line: Feature name[STRING] , Start(BP)[INTEGER] , End(BP)[INTEGER] , Colour(HEX Code)[STRING]
OUTPUT
- plasmid_XXXXXX.svg (outfile) [FILE]
Vector graphic file containing the plasmid map. Files can be viewed and edited in third-party vector graphic software (such as adobe illustrator or Inkscape)
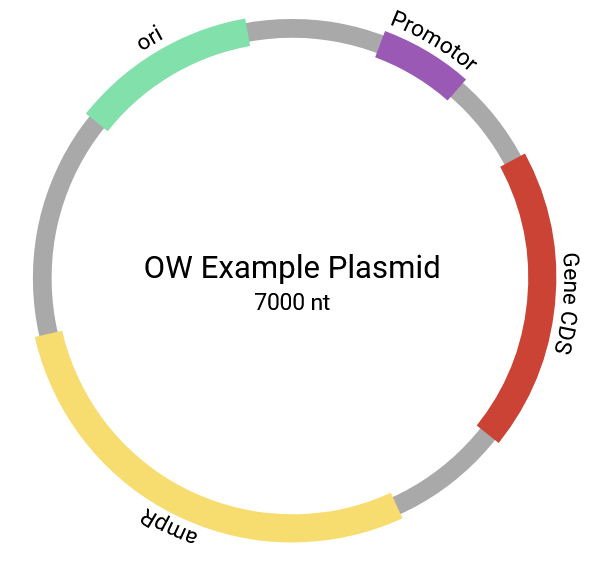
Example Plasmid map
The tool returns a simple plasmid map as a vector graphic. The backbone is indicated by a grey cirlce. Features are placed around the circle in the user specified colours. Feature labels are centered over the featuer and wrap around the plasmid. The center shows the plasmid name and total length. All elements can be edited in compatible vector graphic software.
List of Nucleotides and Terminal Modifications
PLACEHOLDER
- 3' Modifications (three_prime) [STRING]
- 5' Modifications (five_prime) [STRING]
| Letter Code | Full Name |
|---|---|
| OH | Hydroxy |
| P | Monophosphate |
| PPP | Triphosphate |
| FAM | Fluorescein |
| CY3 | Cyanine 3 |
| CY5 | Cyanine 5 |
| BQ1 | Black Hole Quencher 1 |
| BQ2 | Black Hole Quencher 2 |
| 3N7 | 3'-Amino-Modifier C7 |
| 3N3 | 3'-PT-Amino-Modifier C3 |
| 3N6 | 3'-PT-Amino-Modifier C6 |
| 3S3 | 3'-Thiol-Modifier C3 S-S |
| 3S6 | 3'-Thiol-Modifier 6 S-S |
| Letter Code | Full Name |
|---|---|
| OH | Hydroxy |
| P | Monophosphate |
| PPP | Triphosphate |
| FAM | Fluorescein |
| CY3 | Cyanine 3 |
| CY5 | Cyanine 5 |
| BQ1 | Black Hole Quencher 1 |
| BQ2 | Black Hole Quencher 2 |
| 5N5 | 5'-Amino-Modifier 5 |
| 5N6 | 5'-Amino-Modifier C6 |
| N12 | 5'-Amino-Modifier C12 |
| NTT | 5'-Amino-Modifier TEG |
| NTP | 5'-Amino-Modifier TEG PDA |
| NPD | 5'-Amino-Modifier C12-PDA |
| 5NO | 5'-Aminooxy-Modifier-11 |
| 5C5 | 5'-Carboxy-Modifier C5 |
| C10 | 5'-Carboxy-Modifier C10 |
| 5SH | 5'-Thiol-Modifier C6 |
| 5SS | Thiol-Modifier C6 S-S |
| HEX | 5' Hexynyl |
| MAL | Maleimide |
| CHL | TEG-Cholesteryl |